 |
'Light enters, then a miracle happens, and good things come out!' (Quirk & Whelan, 2011*)
|
|
|
|
I'm occasionally asked to investigate weird interventions for children's neurodevelopmental conditions, and recently I've found myself immersed in the world of low-level laser treatments. The material I've dug up is not new - it's been around for some years, but has not been on my radar until now.
A starting point is this 2018 press statement by Erchonia, a firm that makes low-level laser devices for quasi-medical interventions.
They had tested a device that was supposed to reduce irritability in autistic children by applying low-level laser light to the temporal and posterior regions of the head (see Figure 1) for 5 minute sessions twice a week for 4 weeks.
 |
| Figure 1: sites of stimulation by low-level laser |
|
|
The study, which was reported here, was carefully designed as a randomized controlled trial. Half the children received a placebo intervention. Placebo and active laser devices were designed to look identical and both emitted light, and neither the child nor the person administering the treatment knew whether the active or placebo light was being used.
According to Erchonia “The results are so strong, nobody can argue them.” (sic). Alas, their confidence turned out to be misplaced.
The rationale given by Leisman et al (with my annotations in yellow in square brackets) is as follows:
"LLLT promotes cell and neuronal repair (Dawood and Salman 2013) [This article is about wound healing, not neurons] and brain network rearrangement (Erlicher et al. 2002) [This is a study of rat cells in a dish] in many neurologic disorders identified with lesions in the hubs of default mode networks (Buckner et al. 2008)[This paper does not mention lasers]. LLLT facilitates a fast-track wound-healing (Dawood and Salman 2013) as mitochondria respond to light in the red and near-infrared spectrum (Quirk and Whelan 2011*)[review of near-infrared irradiation photobiomodulation that notes inadequate knowledge of mechanisma - see cartoon]. On the other hand, Erlicher et al. (2002) have demonstrated that weak light directs the leading edge of growth cones of a nerve [cells in a dish]. Therefore, when a light beam is positioned in front of a nerve’s leading edge, the neuron will move in the direction of the light and grow in length (Black et al. 2013 [rat cells in a dish]; Quirk and Whelan 2011). Nerve cells appear to thrive and grow in the presence of low-energy light, and we think that the effect seen here is associated with the rearrangement of connectivity."
I started out looking at the registration of the trial on ClinicalTrials.gov. This included a very thorough document that detailed a protocol and analysis plan, but there were some puzzling inconsistencies; I documented them here on PubPeer, and subsequently a much more detailed critique was posted there by Florian Naudet and André Gillibert.
Among other things, there was confusion about where the study was done. The registration document said it was done in Nazareth, Israel, which is where the first author, Gerry Leisman was based. But it also said that the PI was Calixto Machado, who is based in Havana, Cuba.
Elvira Cawthon, from Regulatory Insight, Inc, Tennessee was mentioned on the protocol as clinical consultant and study monitor. The role of the study monitor is specified as follows:
"The study Monitor will assure that the investigator is executing the protocol as outlined and intended. This includes insuring that a signed informed consent form has been attained from each subject’s caregiver prior to commencing the protocol, that the study procedure protocol is administered as specified, and that all study evaluations and measurements are taken using the specified methods and correctly and fully recorded on the appropriate clinical case report forms."
This does not seem ideal, given that the study monitor was in Tennessee, and the study was conducted in either Nazareth or Havana. Accordingly, I contacted Ms Cawthon, who replied:
"I can confirm that I performed statistical analysis on data from the clinical study you reference that was received from paper CRFs from Dr. Machado following completion of the trial. I was not directly involved in the recruitment, treatment, or outcomes assessment of the subjects whose data was recorded on those CRFs. I have not reviewed any of the articles you referenced below so I cannot attest to whether the data included was based on the analyses that I performed or not or comment on any of the discrepancies without further evaluation at this time."
I had copied Drs Leisman and Machado into my query, and Dr Leisman also replied.
He stated:
"I am the senior author of the paper pertaining to a trial of low-level laser therapy in autism spectrum disorder.... I take full responsibility for the publication indicated above and vouch for having personally supervised the implementation of the project whose results were published under the following citation:
Leisman, G. Machado, C., Machado, Y, Chinchilla-Acosta, M. Effects of Low-Level Laser Therapy in Autism Spectrum Disorder. Advances in Experimental Medicine and Biology 2018:1116:111-130. DOI:10.1007/5584_2018_234. The publication is referenced in PubMed as: PMID: 29956199.
I hold a dual appointment at the University of Haifa and at the University of the Medical Sciences of Havana with the latter being "Professor Invitado" by the Ministry of Health of the Republic of Cuba. Ms. Elvira Walls served as the statistical consultant on this project."
However, Dr Leisman denied any knowledge of subsequent publications of follow-up data by Dr Machado. I asked if I could see the data from the Leisman et al study, and he provided a link to a data file on ResearchGate, the details of which I have put on PubPeer.
Alas, the data were amazing, but not in a good way. The main data came from five subscales of the Aberrant Behavior Checklist (ABC)**, which can be combined into a Global score. (There were a handful of typos in the dataset for the Global score, which I have corrected in the following analysis). For the placebo group, 15 of 19 children obtained exactly the same global score on all 4 sessions. Note that there is no restriction of range for this scale: reported scores range from 9 to 154. This pattern was also seen in the five individual subscales. You might think that is to be expected if the placebo intervention is ineffective, but that's not the case. Questionnaire measures such as that used here are never totally stable. In part this is because children's behaviour fluctuates. But even if the behaviour is constant, you expect to see some variability in responses, depending on how the rater interprets the scale of measurement. Furthermore, when study participants are selected because they have extreme scores on a measure, the tendency is for scores to improve on later testing - a phenomenon known as regression to the mean, Such unchanging scores are out of line with anything I have ever come across in the intervention literature. If we turn to the treated group, we see that 20 of 21 children showed a progressive decline in global scores (i.e. improvement), with each measurement improving from the previous one over 4 sessions. This again is just not credible because we'd expect some fluctuation in children's behaviour as well as variable ratings due to error of measurement. These results were judged to be abnormal in a further commentary by Gillibert and Naudet on PubPeer. They also noted that the statistical distribution of scores was highly improbable, with far more even than odd numbers.
Although Dr Machado has been copied into my correspondence, he has not responded to queries. Remember, he was PI for the study in Cuba, and he is first author on a follow-up study from which Dr Leisman dissociated himself. Indeed, I subsequently found that there were no fewer than three follow-up reports, all appearing in a strange journal whose DOIs did not appear to be genuine:
Machado, C., Machado, Y., Chinchilla, M., & Machado, Yazmina. (2019a). Follow-up assessment of autistic children 6 months after finishing low lever (sic) laser therapy. Internet Journal of Neurology, 21(1). https://doi.org/10.5580/IJN.54101 (available from https://ispub.com/IJN/21/1/54101).
Machado, C., Machado, Y., Chinchilla, M., & Machado, Yazmina. (2019b). Twelve months follow-up comparison between autistic children vs. Initial placebo (treated) groups. Internet Journal of Neurology, 21(2). https://doi.org/10.5580/IJN.54812 (available from https://ispub.com/IJN/21/2/54812).
Machado, C., Machado, Y., Chinchilla, M., & Machado, Yazmina. (2020). Follow-up assessment of autistic children 12 months after finishing low lever (sic) laser therapy. Internet Journal of Neurology, 21(2). https://doi.org/10.5580/IJN.54809 (available from available from https://ispub.com/IJN/21/2/54809)
The 2019a paper starts by talking of a study of anatomic and functional brain connectivity in 21 children, but then segues to an extended follow-up (6 months) of the 21 treated and 19 placebo children from the Leisman et al study. The Leisman et al study is mentioned but not adequately referenced. Remarkably, all the original participants participated in the follow-up. The same trend as before continued: the placebo group stagnated, whereas the treated group continue to improve up to 6 months later, even though they received no further active treatment after the initial 4 week period. The 2020 Abstract reported a further follow-up to 12 months. The huge group difference was sustained (see Figure 2). Three of the treated group were now reported as scoring in the normal range on a measure of clinical impairment.
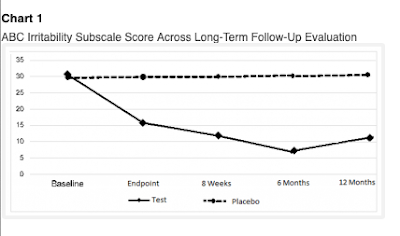 |
Figure 2. Chart 1 from Machado et al 2020
|
In the 2019b paper, it is reported that, after the stunning success of the initial phase of the study, the placebo group were offered the intervention, and all took part, whereupon they proceeded to make an almost identical amount of remarkable progress on all five subscales, as well as the global scale (see Figure 3). We might expect the 'baseline' scores of the cross-over group to correspond to the scores reported at the final follow-up (as placebo group prior to cross-over) but they don't.
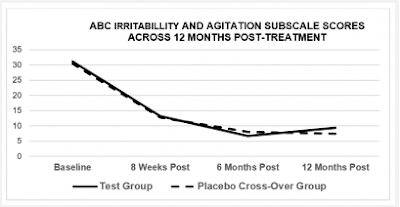 |
Figure 3: Chart 2 of Machado et al 2019b
|
I checked for other Erchonia studies on clinicaltrials.gov. Another study, virtually identical except for the age range, was registered in 2020 with Dr Leon Morales-Quezada of Spaulding Rehabilitation Hospital, Boston as Principal Investigator. Comments in the documents suggest this was conducted after Erchonia failed to get the desired FDA approval. Although I have not found a published report of this second trial, I found a recruitment advertisement, which confusingly cites the NCT registration number of the 2013 study. Some summary results are included on clinicaltrials.gov, and they are strikingly different from the Leisman et al trial, with no indication of any meaningful difference between active and placebo groups in the final outcome measure, and both groups showing some improvement. I have requested fuller data from Elvira Cawthon (listed as results point of contact) with cc. to Dr Morales-Quezada and will update this post if I hear back.
It would appear that at one level this is a positive story, because it shows the regulatory system working. We do not know why FDA rejected Erchonia's request for 510k Market Clearance, but the fact that they did so might indicate that they were unimpressed by the data provided by Leisman and Machado. The fact that Machado et al reported their three follow-up studies in what appears to be an unregistered journal suggests they had difficulty persuading regular journals that the findings were legitimate. If eight 5-minute sessions with a low-level laser pointed at the head really could dramatically improve the function of children with autism 12 months later, one would imagine that Nature, Cell and Science would be scrambling to publish the articles. On the other hand, any device that has the potential to stimulate neuronal growth might also ring alarm bells in terms of potential for harm.
Use of low-level lasers to treat autism is only part of the story. Questions remain about the role of Regulatory Insight, Inc., whose statistician apparently failed to notice anything strange about the data from the first autism study. In another post, I plan to look at cases where the same organisation was involved in monitoring and analysing trials of Erchonia laser devices for other conditions such as cellulite, pain, and hearing loss.
Notes
* Quirk, B. J., & Whelan, H. T. (2011). Near-infrared irradiation photobiomodulation: The need for basic science. Photomedicine and Laser Surgery, 29(3), 143–144. https://doi.org/10.1089/pho.2011.3014. This article states "clinical uses of NIR-PBM have been studied in such diverse areas as wound healing, oral mucositis, and retinal toxicity. In addition, NIR-PBM is being considered for study in connection with areas such as aging and neural degenerative diseases (Parkinson's disease in particular). One thing that is missing in all of these pre-clinical and clinical studies is a proper investigation into the basic science of the NIR-PBM phenomenon. Although there is much discussion of the uses of NIR, there is very little on how it actually works. As far as explaining what really happens, we are basically left to resort to saying 'light enters, then a miracle happens, and good things come out!' Clearly, this is insufficient, if for no other reason than our own intellectual curiosity."
**Aman, M. G., Singh, N. N., Stewart, A. W., & Field, C. J. (1985).
The aberrant behavior checklist: A behavior rating scale for the
assessment of treatment effects. American Journal of Mental Deficiency,
89(5), 485–491. N. B. this is different from the Autism Behavior
Checklist which is a commonly used autism assessment.























